
2018
Understanding disordered structures. — Our research group is involved in the investigation of short range order of liquids, amorphous materials and disordered crystals. We combine experimental data, such as X-ray and neutron diffraction structure factors and EXAFS spectra, with computer modeling tools, such as Reverse Monte Carlo (RMC) and molecular dynamics (MD) simulations. As a result of such an approach, large configurations (tipically tens of thousands of atoms) are provided that are energetically reasonable and consistent (within errors) with experimental data. These configurations are then subjected to various geometrical analyses, so that specific questions concerning the structure of a material may be answered. The group is also responsible for the maintenance and operation of the MTEST neutron diffractometer installed at the 10 MW Budapest Research Reactor. Below we provide some selected results from the year of 2018.
Covalent glasses. — Short range order of As40-xCuxTe60 (x = 0, 10, 20, 25, 30) glasses has been studied by neutron- and X-ray diffraction, combined with extended X-ray absorption fine structure (EXAFS) measurements at the K-edges of all components. Large-scale structural models have been generated by fitting the experimental datasets simultaneously in the framework of the reverse Monte Carlo simulation technique. These simulations revealed that As and Te atoms bind to about 3 and 2 As/Te neighbors, respectively, both of which possess Cu neighbors.
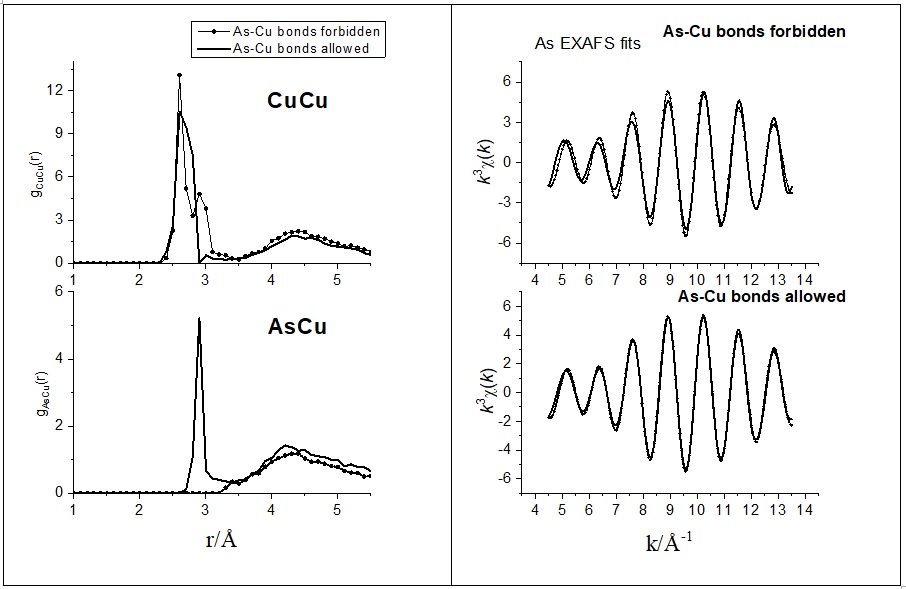
Figure 1. Comparison of As-Cu and Cu-Cu partial pair correlation functions (left) and As K-edge EXAFS fits (right) of glassy As20Cu20Te60 obtained with and without As-Cu bonds
The presence of Cu-As bonds was proven by dedicated simulation runs. In Figure 1, we compare the Cu-Cu and Cu-As partial pair correlation functions and the As20Cu20Te60 As K-edge fits obtained with and without Cu-As bonds. Two observation can be made here: i) the fit quality worsens upon eliminating Cu-As bonding, ii) a secondary Cu-Cu peak appears in the Cu-Cu partial pair correlation function to compensate for the lack of the Cu-As peak. This secondary Cu-Cu peak is necessary to maintain at least the Cu K-edge EXAFS fit quality. The presence of As-Cu bonds was verified by dedicated simulation runs. The Cu-Te bond length is 2.57±0.02Å while the Cu-As distance is as high as 2.86±0.04Å. The results further showed that besides As and Te, Cu atoms also bind to Cu. The total coordination number of Cu is significantly higher than 4 for x = 25 and 30.
Temperature dependent structure and dynamics of ethanol-water mixtures at low alcohol contents. – By making use of literature X-ray diffraction data, extensive molecular dynamics computer simulations have been conducted for ethanol-water liquid mixtures in the water-rich side of the composition range, with 10, 20 and 30 mol % of the alcohol, at temperatures between room temperature and the experimental freezing point of the given mixture. All-atom type (OPLS) interatomic potentials have been assumed for ethanol, in combination with two kinds of rigid water models (SPC/E and TIP4P/2005). Both combinations have provided excellent reproductions of the experimental X-ray total structure factors at each temperature; this provided a strong basis for further structural analyses. Beyond partial radial distribution functions, various descriptors of hydrogen bonded assemblies, as well as of the hydrogen bonded network have been determined from the simulated particle configurations. A clear tendency was observed towards that an increasing proportion of water molecules participate in hydrogen bonding with exactly 2 donor- and 2 acceptor sites as temperature decreases. Concerning larger assemblies held together by hydrogen bonding, the main focus was put on the properties of cyclic entities: it was found that, similarly to methanol-water mixtures, the number of hydrogen bonded rings has increased with decreasing temperature. However, for ethanol-water mixtures the dominance of not the six-, but of the five-fold rings could be observed (see Figure 2).
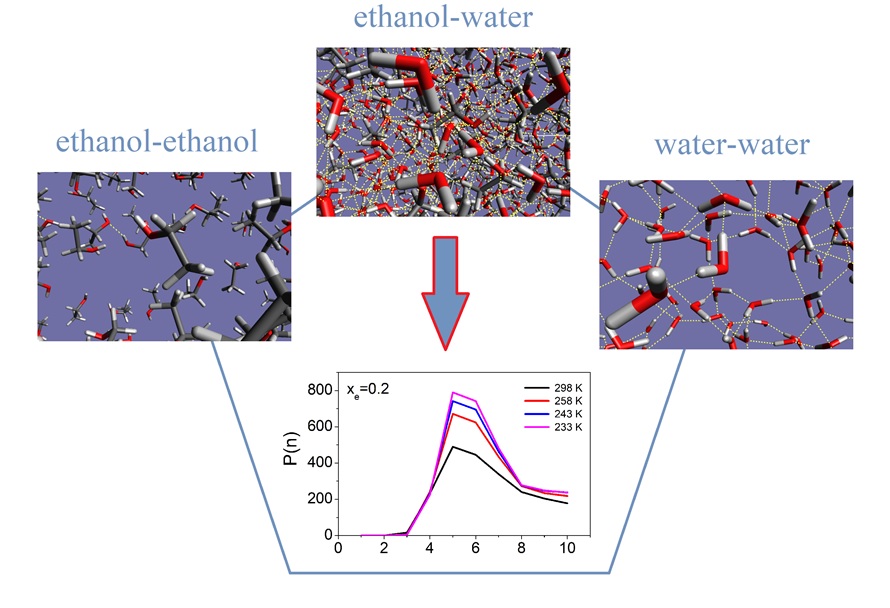
Figure 2. Temperature dependence of hydrogen bond ring size distribution in water-20 mol% ethanol mixture
Starting from the molecular dynamics simulations mentioned above, we took a closer look at the time-dependent behavior of molecules. Temperature dependent hydrogen bond energetics and dynamical features, such as the diffusion coefficient and reorientational times, have been determined for ethanol-water mixtures with 10, 20 and 30 mol % of ethanol. Concerning pairwise interaction energies between molecules, it was found that water-water interactions become stronger, while ethanol-ethanol ones become significantly weaker in the mixtures, than the corresponding values characteristic to the pure substances. Concerning the diffusion processes, for all concentrations the activation barrier of water and ethanol molecule become very similar to each other. Reorientational motions of water and ethanol become slower as ethanol concentration is increasing. Characteristic reorientational times of water in the mixtures are substantially longer than these values in the pure substance. On the other hand, for ethanol this change is only moderate. Reorientational motions of water (especially the ones related to the H-bonded interaction) become very similar for those of ethanol in the mixtures.
The structure of the simplest liquid aldehydes. – Although aliphatic aldehydes (a.k.a. alkanals, compounds with chain-end –CHO groups) constitute an essential group of organic substances, structural studies of them are scarce. Synchrotron X-ray diffraction experiments and molecular dynamics simulations have been performed on simple aliphatic aldehydes in the liquid state, from propanal to nonanal. The performance of the OPLS all-atom interaction potential model for aldehydes has been assessed via direct comparison of simulated and experimental total scattering structure factors. In general, MD results reproduce the experimental data at least semi-quantitatively. However, a slight mismatch can be observed between the two datasets in terms of the position of the main diffraction maxima. Partial radial distribution functions (PRDF) have also been calculated from the simulation results. Clear differences could be detected between the various O-H partial radial distribution functions, depending on whether the H atom is attached to the carbon atom that is doubly bonded to the oxygen atom of the aldehyde group or not. Based on the 3 different O-H PRDF-s, as well as on the various H-H PRDF-s, it may be suggested that neighboring molecules turn toward each other (somewhat) preferentially by their aldehyde ends. From gOO(r) and gC’C’(r), and from intermolecular angular correlations presented in Figure 5, it may be discerned that no (or at most, extremely weak) orientational correlations are present between neighboring aldehyde groups.
As a follow-up of the above series of experiments, the total scattering structure factors of pure liquid n-pentanol, pentanal, and 5 of their mixtures have been determined by high energy synchrotron X-ray diffraction experiments. For the interpretation of measured data, molecular dynamics computer simulations were performed, utilizing ‘all-atom’ type force fields. The diffraction signals in general resemble each other over most of the monitored scattering variable, Q, range above 1 Å-1, but the absolute values of the intensities of the small-angle scattering maximum (‘pre-peak’, ‘first sharp diffraction peak’), around 0.6 Å-1, change in an unexpected fashion, non-linearly with the composition. MD simulations are not able to reproduce this low-Q behavior; on the other hand, they do reproduce the experimental diffraction data above 1 Å-1 rather accurately. Partial radial distribution functions are calculated based on the atomic coordinates in the simulated configurations. Inspection of the various O-O and O-H partial radial distribution functions clearly shows that both the alcoholic and the aldehydic oxygens form hydrogen bonds with the hydrogen atoms of the alcoholic OH-group.
Aqueous salt solutions. –Highly concentrated aqueous lithium chloride solutions have been investigated by classical molecular dynamics (MD) and reverse Monte Carlo (RMC) simulations. At first MD calculations have been carried out applying twenty-nine combinations of ion-water interaction models at four salt concentrations. The structural predictions of the different models have been compared, the contributions of different structural motifs to the partial pair correlation functions (PPCF) have been determined. Particle configurations obtained from MD simulations have been further refined using the RMC method to get better agreement with experimental X-ray and neutron diffraction data. The PPCFs calculated from MD simulations have been fitted together with the experimental structure factors to construct structural models that are as consistent as possible with both the experimental results and the results of the MD simulations. The MD models have been validated according to the quality of the fits. Although none of the tested MD models can describe the structure perfectly at the highest investigated concentration, their comparison made it possible to determine the main structural properties of that solution as well. It was found that four nearest neighbors (oxygen atoms and chloride ions together) are around a lithium ion at each concentration, while in the surroundings of the chloride ion hydrogen atom pairs are replaced by one lithium ion as the concentration increases. While in pure liquid water four water molecules can be found around a central water molecule, near the solubility limit nearly all water molecules are connected to two chloride ions (via their hydrogen atoms) and one lithium ion (by their oxygen atoms).
